摘要:本文对茶叶中的11种有机氯和拟除虫菊酯类农药残留量进行了同时快速分析。通过简单的样品前处理方法,将茶样与吸附剂充分研磨后,进行柱层析净化预分离,最后经气相色谱检测。文中分别对吸附剂、层析柱和洗脱剂等条件进行了详细考察,优化的方法为:1.0 g茶样浸润后与4.0 g中性Al2O3吸附剂充分研磨,混合物装入预填2.0 g无水Na2SO4+2.0 g Florisil+0.1 g活性炭的自制层析柱净化预分离,以20 mL乙酸乙酯:正己烷(2:3,V/V)洗脱;该法线性范围为0.1~2.0 mg/L,相关系数(r)大于0.9962(除了氟氯氰菊酯和氯氰菊酯),检出限(LOD)在0.0000321~0.1451379 ng之间,样品加标回收率(R)在68.44%~119.20%,不同加标级别回收率结果的相对标准偏差值(RSD)均低于14.73%。此外,对茶样的基质干扰(ME)问题进行了详细讨论,并采用基质匹配校准曲线法(MC)有效解决ME问题。经实验验证,该方法快速、准确、简便、环保,适用于茶叶中农残的同时、快速分离与分析。
关键词:农残分析;茶叶;样品前处理方法;基质干扰
农药残留问题一直是影响我国茶叶出口的主要贸易壁垒。世界上已有18个国家和组织颁发了349项农药允许残留标准,并且一些茶叶主要进口国还对茶叶中的农药残留量制定了更为严格的限量法规[1],使我国茶叶出口面临巨大压力。茶叶中含有大量的特有化学成分,如茶色素、茶多酚、咖啡碱、茶碱、茶氨酸和茶多糖等等,在进行农残分析过程中,茶样中有94%以上的物质会作为共存杂质而对分析造成严重的响应干扰,影响结果的准确性,甚至出现假阳性结果。因此,茶叶样品较一般的蔬菜水果而言,其基质更为复杂,基质干扰(Matrix effects,ME)[2,3]问题尤为突出。
目前茶叶农残分析的样品前处理方法主要有:溶剂萃取法(SE)[4,5]、固相萃取法(SPE)[4~6]、凝胶渗透色谱法(GPC)[7]、加速溶剂萃取方法(ASE)[7,8]、固相微萃取技术(SPME)[9]、搅拌棒吸附萃取方法(SBSE)[10]、液相微萃取(LPME)[11,12]和QuEChERS 法[13]等等。SE法基质干扰问题较为严重;SPE法成本较高;GPC法费时且回收率较低,有机溶剂消耗量大;ASE法必需进行后续的净化处理;SPME法成本较高,测样重复性、重现性均较差;SBSE法操作复杂且成本较高;LPME法灵敏度和准确度相对较低,样品基质的影响较大;QuEChERS法易产生基质干扰,影响检测结果的准确度与可靠性。因此,有必要建立一种简单、快速、高效、环保的方法,对茶样进行前处理。
本文以常见的几种有机氯和拟除虫菊酯农药为分析目标物,建立了一种简单的茶叶样品前处理方法,将干茶加水浸润后直接与优选的吸附剂混合研磨,最终得到均匀分散的混合物,装入已预先填有一定吸附剂和除水剂的层析柱,然后用混配的有机溶剂洗脱,洗脱液浓缩后可直接进行仪器分析。同时,本文对该样品前处理方法的各影响因素进行了优化;对该方法的 ME问题进行考察;最后从线性关系、检出限(LOD)、加标回收率及相对标准偏差(RSD)等指标对该样品前处理方法进行了可行性评价。
1.1 仪器和试剂
Agilent 6890N气相色谱仪(美国Agilent公司),配备电子捕获检测器(ECD);Agilent 7890B-7000C气相色谱-串联质谱仪(美国Agilent公司),配备7693自动进样器;HH数显恒温水浴锅(金坛市金城国胜仪器厂);AUW220D电子天平(日本 Shimadzu公司);SB3200D超声波清洗机(宁波新芝生物科技有限公司);JYL-A100料理机(九阳股份有限公司)、SX2-4-10箱式电阻炉(上海实验仪器厂有限公司)
农残级正己烷和乙酸乙酯、色谱级甲醇均购自百灵威科技有限公司进口分装;分析纯乙酸乙酯、分析纯二氯甲烷购自天津市科密欧化学试剂有限公司;分析纯丙酮(国药集团化学试剂有限公司);分析纯无水Na2SO4(天津市恒兴化学试剂制造有限公司);Florisil硅藻土(60~100目,250 g)、活性炭颗粒(1000 g)和硅胶(60~100目,500 g)均购自国药集团化学试剂有限公司;C18 (40~60 μm,100 g,北京艾杰尔科技有限公司);柱层析用中性Al2O3(100~200目,500 g,上海陆都化学试剂厂)。
联苯菊酯(96.0%,100 mg)、氟氯氰菊酯(94.2%,10 mg)、氯氰菊酯(97.1%,10 mg)、p,p’-DDD(98%,10 mg)、α-硫丹(97.5%,10 mg)、β-硫丹(99.9%,10 mg)均购自美国 AccuStandard®,Inc.;p,p′-DDT(99.7%,0.1 g)、三氟氯氰菊酯(98.0%,0.1 g)、溴氰菊酯(99.5%,0.25 g)、顺式氰戊菊酯(98.0%,0.25 g)购自德国 Dr. Ehrenstorfer. GmbH;δ-BHC为 1.0 mL(100 μg/mL),购自农业部环境保护科研监测所。
1.2 试剂预处理
无水Na2SO4:于650 ℃马弗炉中灼烧4 h,密封放置于干燥器中冷却至室温备用;中性 Al2O3:于550 ℃马弗炉中灼烧4 h,密封放置于干燥器内,冷却至室温。使用前130 ℃活化5 h,可保存一周,过期重新活化;Florisil硅藻土:100~200目,于650 ℃马弗炉中烘烤4 h,密封放置于干燥器内,冷却至室温,使用前加5%超纯水去活;硅胶:于130 ℃马弗炉中烘烤2 h密封放置于干燥器内,冷却至室温,备用。
1.3 GC-ECD检测条件
DB-5色谱柱(30.0 m×0.25 mm×0.25 μm,美国安捷伦科技公司);进样口温度 290 ℃;检测器温度300 ℃;不分流进样;载气为高纯氮气;柱流量为1.0 mL/min;柱前压为103 kPa;进样量为1 μL;外标法定量。升温程序:
1.4 样品前处理方法
内径1.0 cm的自制玻璃层析柱中,依次填入2.00 g处理过的Florisil硅藻土和0.10 g活性炭颗粒,备用。茶样粉碎后过20目筛,准确称取1.00 g处理过的茶样于玻璃研钵中,加入0.5 mL超纯水浸润,再加入4.00 g中性Al2O3吸附剂,充分研磨至均匀分散,将混合物彻底转移至上述层析柱内,轻敲柱壁填实,上端压入适量脱脂棉,倒入20 mL乙酸乙酯:正己烷(2:3,V/V)混合溶剂,加压洗脱至溶液完全流出,收集洗脱液,40 ℃下水浴,恒温氮吹浓缩至近干,以正己烷置换溶剂并定容至1.00 mL,待仪器检测。
1.5 标准溶液的配制
1.5.1 SC法标准溶液
五种有机氯混标:分别移取适量的 p,p'-滴滴涕(p,p'-DDT)、p,p′-滴滴滴(p,p′-DDD)、α-硫丹、β-硫丹和δ-六六六(δ-BHC)于5 mL容量瓶中,正己烷定容得浓度为5 mg/L的有机氯混标A。
六种拟除虫菊酯混标:分别移取适量的氯氰菊酯、氟氯氰菊酯、联苯菊酯、顺式氰戊菊酯、溴氰菊酯和三氟氯氰菊酯于5 mL容量瓶中混合,正己烷定容得浓度为5 mg/L的拟除虫菊酯混标B。
移取适量上述溶液A和B混合,以正己烷稀释至所需浓度,即得纯溶剂校准曲线法(Solvent calibration,SC)的系列混标。
1.5.2 MC法标准溶液
筛选经国标方法[5]检测,不含此11种有机氯和拟除虫菊酯农残的茶样作为基质匹配校准曲线法(Matrix-matched calibration,MC)的匹配基质[2,3],同上述1.4方法进行样品前处理后,所得的正己烷溶液即为样品空白(Sample blank,SB)。配制MC法系列混标时,根据浓度需要,分别移取适量体积的混标A和B于同一个容量瓶,以样品空白稀释定容。
2.1 研磨提取方法优化
采用自制的内径1.0 cm玻璃层析柱,参考文献[14]的方法,自下而上依次预填2.0 g无水Na2SO4、2.0 g Florisil硅藻土、1.0 g中性Al2O3,备用。准确称取1.00 g粉碎的茶样,平行4份,每份茶样均加标0.5 mg/L,加入0.5 mL超纯水浸润后,分别与4.00 g的不同吸附剂(Florisil硅藻土、中性Al2O3、硅胶或C18)充分研磨,得均匀分散的混合物,装柱,轻敲柱壁填实,上端压入适量脱脂棉。量取30 mL正己烷:乙酸乙酯(95:5,V/V)混合溶剂[14],取少量彻底转移研钵中的混合物后,剩余溶剂倾入层析柱,加压洗脱至溶液完全流出,收集洗脱液,40 ℃下水浴,恒温氮吹浓缩至近干,以正己烷置换溶剂并定容至1.00 mL,待仪器检测,SC法定量。
表1 不同吸附剂种类和不同用量条件下11种农药的加标回收率R
Table 1 Recoveries of 11 pesticides using various types of adsorbent at different dosages
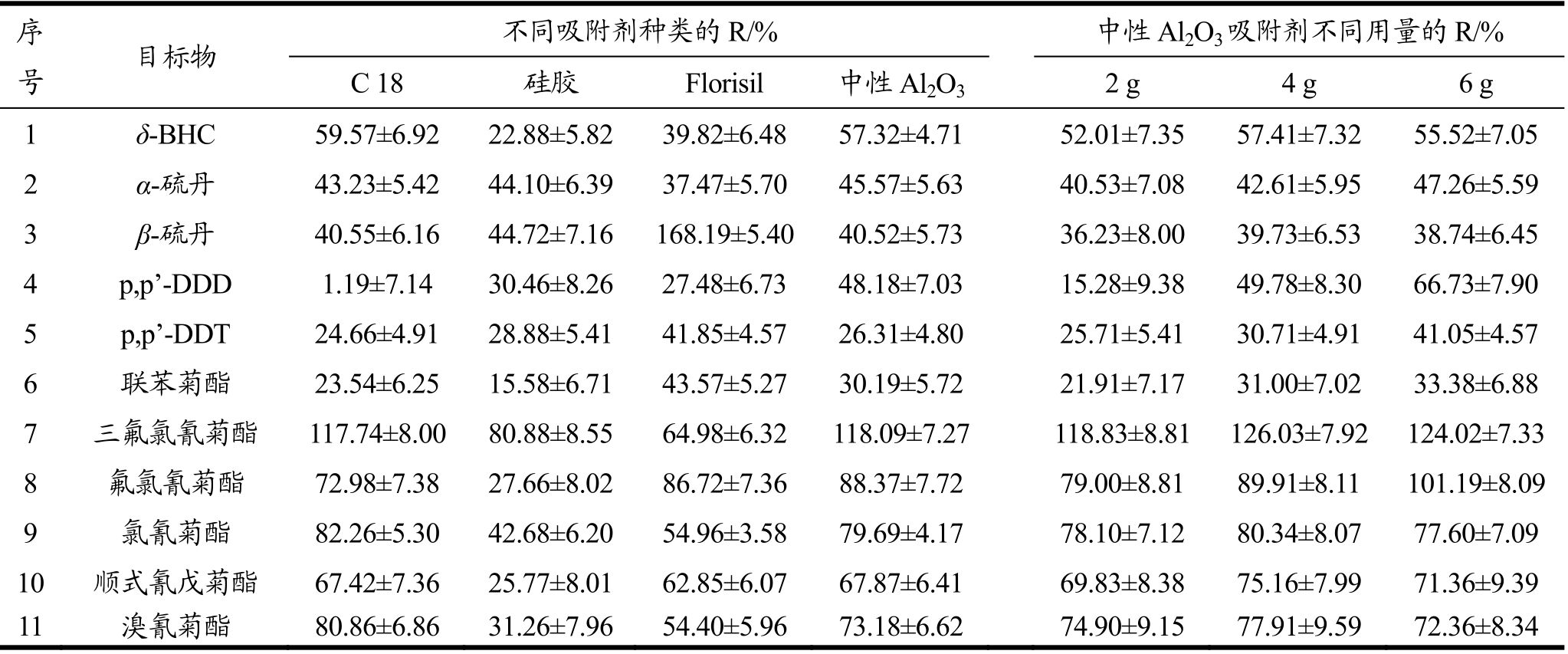
注:氟氯氰菊酯和氯氰菊酯的标准品在GC-ECD检测中出现多个异构体峰,故以其峰面积之和来定量。
序号 目标物 不同吸附剂种类的R/% 中性Al2O3吸附剂不同用量的R/% C 18 硅胶 Florisil 中性Al2O32 g 4 g 6 g 1 δ-BHC 59.57±6.92 22.88±5.82 39.82±6.48 57.32±4.71 52.01±7.35 57.41±7.32 55.52±7.05 2 α-硫丹 43.23±5.42 44.10±6.39 37.47±5.70 45.57±5.63 40.53±7.08 42.61±5.95 47.26±5.59 3 β-硫丹 40.55±6.16 44.72±7.16 168.19±5.40 40.52±5.73 36.23±8.00 39.73±6.53 38.74±6.45 4 p,p’-DDD 1.19±7.14 30.46±8.26 27.48±6.73 48.18±7.03 15.28±9.38 49.78±8.30 66.73±7.90 5 p,p’-DDT 24.66±4.91 28.88±5.41 41.85±4.57 26.31±4.80 25.71±5.41 30.71±4.91 41.05±4.57 6 联苯菊酯 23.54±6.25 15.58±6.71 43.57±5.27 30.19±5.72 21.91±7.17 31.00±7.02 33.38±6.88 7 三氟氯氰菊酯 117.74±8.00 80.88±8.55 64.98±6.32 118.09±7.27 118.83±8.81 126.03±7.92 124.02±7.33 8 氟氯氰菊酯 72.98±7.38 27.66±8.02 86.72±7.36 88.37±7.72 79.00±8.81 89.91±8.11 101.19±8.09 9 氯氰菊酯 82.26±5.30 42.68±6.20 54.96±3.58 79.69±4.17 78.10±7.12 80.34±8.07 77.60±7.09 10 顺式氰戊菊酯 67.42±7.36 25.77±8.01 62.85±6.07 67.87±6.41 69.83±8.38 75.16±7.99 71.36±9.39 11 溴氰菊酯 80.86±6.86 31.26±7.96 54.40±5.96 73.18±6.62 74.90±9.15 77.91±9.59 72.36±8.34
由表1的加标回收率(Recovery,R)结果可见,中性Al2O3并非对所有目标物效果最佳,但考虑其本身的性质、研磨效果(茶样与吸附剂的研磨颗粒细腻且均匀)以及洗脱液的颜色情况(洗脱液澄清透明且无色),选择中性Al2O3作为研磨吸附剂。
采用不同量的中性Al2O3作为研磨吸附剂进行同上前处理,由表1的加标回收率结果可见,使用4.0 g的中性Al2O3研磨后,绝大部分目标物的加标回收率较高,且与使用6.0 g中性Al2O3研磨的回收率结果相近,故中性Al2O3吸附剂的用量确定为4.0 g。
2.2 柱层析净化方法优化
2.2.1 柱层析填料
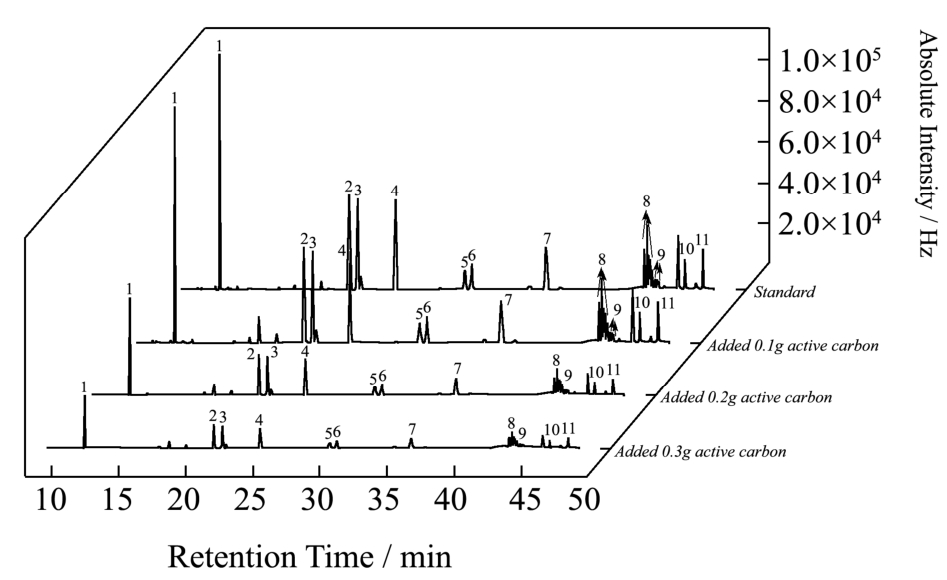
图1 不同量活性炭处理后的11种目标物混标(1.0 mg/L)的色谱图对比
Fig.1 Chromatograms of 11 pesticides absorbed by different amounts of activated carbon
注:1代表δ-BHC;2代表p,p’-DDD;3代表p,p’-DDT;4代表α-硫丹;5代表β-硫丹;6代表联苯菊酯;7代表三氟氯氰菊酯;8代表氟氯氰菊酯;9代表氯氰菊酯;10代表顺式氰戊菊酯;11代表溴氰菊酯。
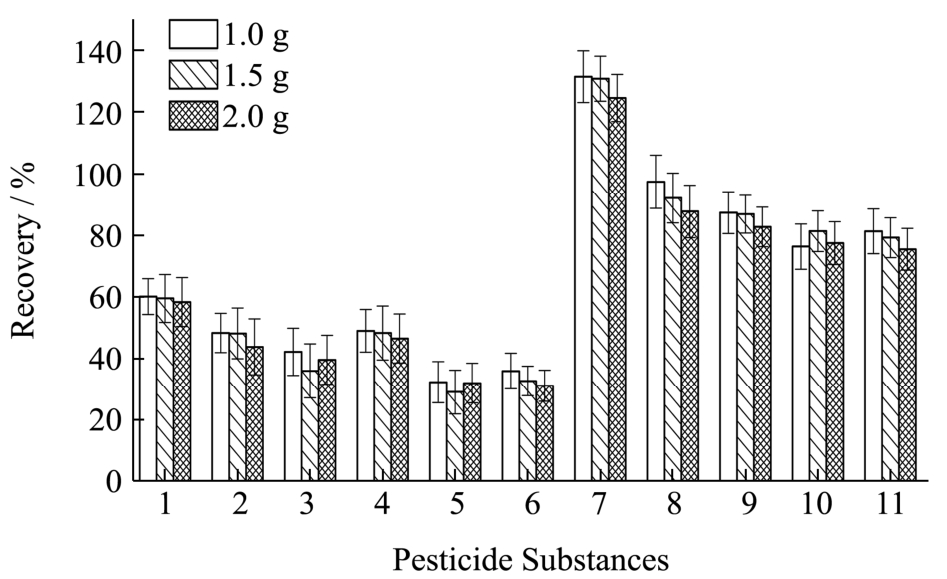
图2 不同Florisil硅藻土用量的柱层析条件下11种目标物的加标回收率
Fig.2 Recoveries of 11 pesticides with different amounts of Florisil adsorbent
注:1代表δ-BHC;2代表p,p’-DDD;3代表p,p’-DDT;4代表α-硫丹;5代表β-硫丹;6代表联苯菊酯;7代表三氟氯氰菊酯;8代表氟氯氰菊酯;9代表氯氰菊酯;10代表顺式氰戊菊酯;11代表溴氰菊酯。氟氯氰菊酯和氯氰菊酯以其异构体的峰面积之和定量。
由于茶叶样品基质复杂,需要在研磨吸附后结合柱层析的方法,进一步净化预分离。因本实验中的研磨吸附剂选用中性 Al2O3,柱层析的填料主要考察:Florisil硅藻土和活性炭颗粒。
活性炭吸附能力强且难于洗脱,为了不造成目标物的吸附损失,在脱除色素的基础上用量越少越好,因此首先开展预试验。参照国标方法[5]处理的茶样提取液浓缩至5.0 mL,分别加入0.1 g、0.2 g和0.3 g活性炭颗粒,振荡10 min后发现,加0.1 g活性炭的溶液呈现浅绿色;加0.2 g活性炭的溶液几乎无色;加0.3 g活性炭的溶液无色。此外,在5.0 mL的11种农药混标(1.0 mg/L)中分别加入0.1 g、0.2 g和0.3 g活性炭,振荡10 min后经GC-ECD检测,对比色谱图(图1)发现,加0.1 g活性炭吸附了极少量的目标物,而加0.2 g或0.3 g活性炭吸附较多的目标物组分,因此考察Florisil硅藻土与0.1 g活性炭颗粒组合使用的净化效果。
在预填2.0 g无水Na2SO4的层析柱内,分别由下至上依次装填:1.0 g Florisil+0.1 g活性炭、1.5 g Florisil+0.1 g活性炭、2.0 g Florisil+0.1 g活性炭,其他步骤同上2.1,加标回收率结果见图2。不同Florisil硅藻土用量对目标物的加标回收率影响不大,但随着Florisil硅藻土用量的增加,洗脱液颜色减淡,基质中的干扰物质减少,故层析柱的装填内容为:2.0 g无水Na2SO4+2.0 g Florisil+0.1 g活性炭+研磨混合物(由下至上)。
2.2.2 洗脱剂配比

图3 不同乙酸乙酯:正己烷的配比为洗脱剂条件下11种目标物的加标回收率
Fig.3 Recoveries of 11 pesticides eluted with ethyl acetate and hexane at different volume ratios
注:1代表δ-BHC;2代表p,p’-DDD;3代表p,p’-DDT;4代表α-硫丹;5代表β-硫丹;6代表联苯菊酯;7代表三氟氯氰菊酯;8代表氟氯氰菊酯;9代表氯氰菊酯;10代表顺式氰戊菊酯;11代表溴氰菊酯。
在不改变其他实验条件的情况下(同2.2.1),分别考察不同体积配比的乙酸乙酯:正己烷混合溶剂,作为洗脱剂,对目标物加标回收率的影响:0:10、1:9、2:8、3:7、4:6、1:1、6:4、7:3和8:2。由图3的曲线走势可见,乙酸乙酯:正己烷的体积比为6:4时,大部分目标物的加标回收率较高,继续增大乙酸乙酯的体积比例对回收率结果影响不大;且该体积配比下的洗脱液呈现无色,随后若增大乙酸乙酯的体积比例,洗脱液颜色加深,干扰物质明显增多。综合考虑两个方面因素,洗脱剂确定为乙酸乙酯:正己烷(6:4,V/V)混合溶剂。
2.2.3 洗脱剂用量
由图4的洗脱曲线可见,对于δ-BHC(编号1)、p,p’-DDD(编号2)和氟氯氰菊酯(编号8),其加标回收率随洗脱剂体积的增加而持续增大;但对于其他目标物的加标回收率,当洗脱剂用量大于25 mL时,变化不大。所以本实验最终确定洗脱剂用量为25 mL,既确保绝大部分目标物充分洗脱,又尽量减小基质干扰的ME问题。


图4 不同洗脱剂体积条件下11种目标物的加标回收率
Fig.4 Recoveries of 11 pesticides eluted with different volumes of eluent
注:1代表δ-BHC;2代表p,p’-DDD;3代表p,p’-DDT;4代表α-硫丹;5代表β-硫丹;6代表联苯菊酯;7代表三氟氯氰菊酯;8代表氟氯氰菊酯;9代表氯氰菊酯;10代表顺式氰戊菊酯;11代表溴氰菊酯。
2.3 基质干扰考察
由于茶样基质复杂,杂质和干扰物质很多,基质干扰(Matrix effect,ME)问题尤为突出。分别按照1.5.1和1.5.2方法配制0.5 mg/L的SC法和MC法混标,经GC-ECD检测,对比色谱图发现,所有目标物都或多或少信号增强,呈现正ME,与文献报导的结果相似[16,17],存在基体诱导色谱响应增强(Matrixinduced chromatographic response enhancement,MCRE)效应。分析其原因可能主要在于:1)茶样本身有机物质组成较水果蔬菜等食品更为复杂,且含量高,引发ME效应的概率很高;2)样品前处理方法的不足,净化过程中不能够彻底分离前一步骤所同时提取的大量干扰物质,导致ME问题。
因此,为提高分析的准确度、灵敏度和选择性,排除ME问题所造成的结果偏高,甚至是“假阳性”结果,在实际样品的检测过程中,采用MC法校准工作曲线(同 1.5.2),并以此定量计算茶样中的农药残留量。
2.4 方法验证
2.4.1 线性与范围
同上1.5.2方法,配制浓度依次为0.1、0.4、0.8、1.2、1.6、2.0 mg/L的MC法混标,进样1.0 μL,经GC-ECD检测,绘制MC法工作曲线,其线性相关系数见表2.。11种目标物在0.1~2.0 mg/L的浓度范围内呈现良好的线性,其线性相关系数值r在0.9962以上,除了氟氯氰菊酯和氯氰菊酯。
2.4.2 方法检出限和定量限
依据IUPAC对于检出限的定义,采用3倍信号(Signal)与噪音(Noise)比值的方法计算方法检出限(Limit of detection,LOD),即3×S/N;采用10×S/N的方法计算方法定量限(Limit of quantification,LOQ)。采用IUPAC推荐的单浓度校正方法,以0.01 mg/L的MC法混标作为标准计算斜率,即S;通过Agilent GC-MS/MS工作站(Agilent MassHunter Workstation)的定量分析软件 Qualitative Analysis B.04.00计算噪音值,即N。据此计算的各目标物的方法LOD和LOQ值见表2。各目标物的LOD值在0.0000321~0.1451379 ng,LOQ 值在 0.0001070~0.4837929 ng,检出限较低。
表2 农药目标物的线性相关系数(r)、方法检出限(LOD)和定量限(LOQ)、茶样不同级别加标的回收率
Table 2 Linear correlation coefficient (r), limit of detection (LOD), and limit of quantification (LOQ) of 11 pesticides and recoveries from tea samples spiked with different concentration of standard

序号 目标物 线性相关系数r LOD/ng LOQ/ng 茶样加标回收率R/%(n=3) RSD/%0.1 mg/L SD 0.5 mg/L SD 1.0 mg/L SD 1 δ-BHC 0.9968 0.0000321 0.0001070 88.86 6.17 95.72 6.23 93.90 5.44 3.83 2 α-硫丹 0.9983 0.0006021 0.0020069 68.44 9.38 74.94 8.34 79.74 7.36 7.63 3 β-硫丹 0.9987 0.0008172 0.0027241 95.24 5.32 105.90 4.52 109.46 3.99 7.15 4 p,p’-DDD 0.9978 0.0001792 0.0005974 72.50 9.88 81.22 6.12 94.96 3.81 13.66 5 p,p’-DDT 0.9962 0.0018430 0.0061434 69.08 6.41 78.38 4.36 81.65 2.56 8.54 6 联苯菊酯 0.9998 0.0001119 0.0003730 72.74 7.00 86.35 5.69 90.93 2.24 11.35 7 三氟氯氰菊酯 0.9989 0.0003880 0.0012934 74.40 8.89 91.10 7.36 96.77 6.85 13.30 8 氟氯氰菊酯 0.9909 0.0015022 0.0050074 89.34 17.30 113.20 12.65 119.20 7.37 14.73 9 氯氰菊酯 0.9954 0.0019079 0.0063598 71.15 13.36 75.92 10.26 87.55 8.42 10.79 10 顺式氰戊菊酯 0.9999 0.0005495 0.0018318 78.00 5.10 83.77 5.25 89.19 3.07 6.69 11 溴氰菊酯 0.9998 0.1451379 0.4837929 92.37 6.27 98.45 3.45 103.14 2.35 5.51
2.4.3 分析空白的控制
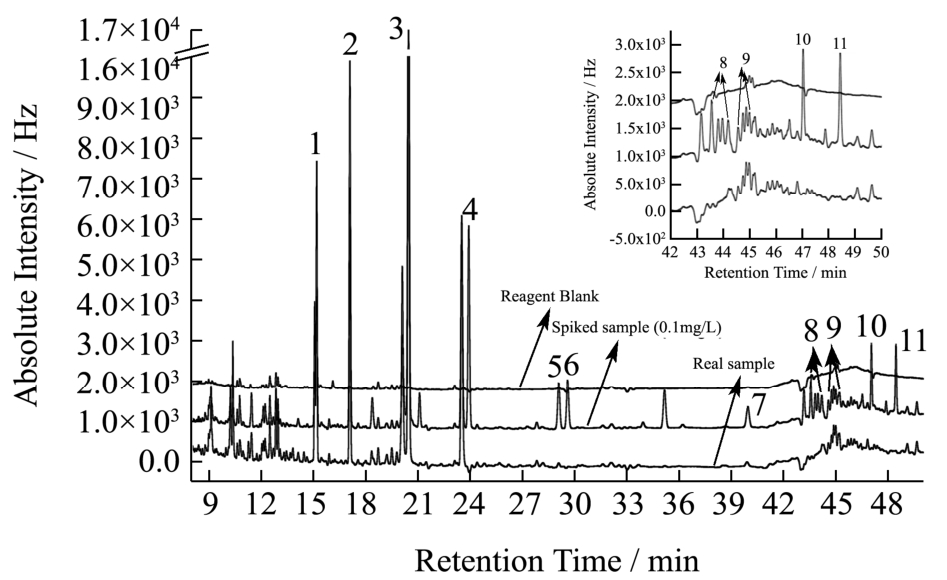
图5 试剂空白、实际茶叶样品和加标0.1 mg/L茶样的GC/ECD色谱图
Fig.5 GC/ECD chromatograms of reagent blank, real tea sample, and spiked tea sample (0.1 mg/L)
注:1代表δ-BHC;2代表p,p’-DDD;3代表p,p’-DDT;4代表α-硫丹;5代表β-硫丹;6代表联苯菊酯;7代表三氟氯氰菊酯;8代表氟氯氰菊酯;9代表氯氰菊酯;10代表顺式氰戊菊酯;11代表溴氰菊酯;Reagent blank为试剂空白;Real sample为实际茶叶样品;Spiked sample为加标0.1 mg/L的茶叶样品。
在试剂空白样品制备的过程中,除了没有茶样,其他操作步骤完全与1.4相同,经GC-ECD检测后,对比试剂空白(Reagent Blank)与茶样(Real Sample)的色谱图(图5)可见,在上述11种目标物的出峰时间段,试剂空白中没有其他物质同时出峰,不存在茶样之外的外源物质干扰问题。
2.4.4 方法的加标回收率
分别对茶样进行0.1、0.5、1.0 mg/L三种不同级别的加标浓度,开展方法的加标回收率实验,每个级别平行三份,静置8 h后,同1.4方法进行样品前处理,进GC-ECD检测。表2结果表明,各目标物的加标回收率R值在68.44%~119.20%之间,目标物的损失在可以接受的范围之内,加标浓度0.1 mg/L的回收率较低,原因在于低的加标量受实验操作过程中的系统误差和偶然误差影响较大;所有目标物的不同浓度加标量的回收率结果之间的RSD值均低于14.73%,符合农残分析的准确度和精密度要求。
2.5 实际样品测定
随机从当地的大型超市购买三种不同茶类的样品:红茶、绿茶和乌龙茶,采用优化后的样品前处理方法(1.4),每个样品的处理平行三份,对其中的上述11种农药残留量情况进行检测。通过数据分析,发现茶样中残留极少量的β-硫丹、p,p’-DDT、联苯菊酯和顺式氰戊菊酯,但其残留量均未超出我国和欧盟的相关限量标准的规定,即未超标。
3.1 本文建立了简单的研磨提取后柱层析净化的样品前处理方法,同时分离分析茶叶中的11种有机氯和拟除虫菊酯农残。通过方法学验证实验表明,所建立的样品前处理方法适用于茶叶样品中的农残分析,较溶剂萃取、超声提取和GPC法等方法[4~7]更为简单易行,避免了大量有机溶剂的使用所造成的危害;且较SPE[4~6]、ASE[7,8]和SPME[9]方法成本低、重现性好,适于普遍应用。
3.2 该样品前处理方法有效去除了茶样基质中的大量干扰物质,解决了基质干扰问题(ME),选择性好,灵敏度高,且快速、准确、简便、环保,适用于在实际生产中推广应用。在后续的研究工作中,我们尝试将主要在以下三个方面深入开展研究:(1)将该处理方法与其他方法的效果进行比较,如国家标准方法(GB/T 23204-2008[5]);(2)将该方法应用于有机氯、拟除虫菊酯、氨基甲酸酯和新烟碱类,不同种类农残的同时分析;(3)对不同类茶样(绿茶、红茶、白茶、黄茶、黑茶和乌龙茶)的基质干扰问题进行考察与比较,从而建立更为有效的样品前处理方法,广泛用于不同茶类中的不同类农残同时测定。
参考文献:
[1] 张顺玮.茶叶中残留啶虫脒测定方法的研究[D].上海:同济大学理学部,2009 ZHANG Shun-wei. Determination method research of acetamiprid residue in tea [D]. Shanghai: School of Sciences in Tongji University, 2009
[2] Stefan Kittlaus, Julia Schimanke, Guenther Kempe, et al. Assessment of sample cleanup and matrix effects in the pesticide residue analysis of foods using postcolumn infusion in liquid chromatography-tandem mass spectrometry [J]. J. Chromatogr. A, 2011, 1218(46): 8399-8410
[3] LI Yan, CHEN Xi, FAN Chun-lin, et al. Compensation for matrix effects in the gas chromatography-mass spectrometry analysis of 186 pesticides in tea matrices using analyte protectants [J]. J. Chromatogr. A, 2012, 1266: 131-142
[4] GB/T 23205-2008,茶叶中448种农药及相关化学品残留量的测定 液相色谱-串联质谱法[S] GB/T 23205-2008, Determination of 448 Pesticides and related chemicals residues in Tea-LC-MS-MS method [S]
[5] GB/T 23204-2008,茶叶中519种农药及相关化学品残留量的测定 气相色谱-质谱法[S] GB/T 23204-2008, Determination of 519 Pesticides and related chemicals residues in Tea-GC-MS method [S]
[6] Claudia Oellig, Wolfgang Schwack. planar solid phase extraction clean-up for pesticide residue analysis in tea by liquid chromatography-mass spectrometry [J]. J. Chromatogr. A, 2012, 1260(19): 42-53
[7] HU Bei-zhen, SONG Wei-hua, XIE Li-ping, et al. Determination of 33 pesticides in tea using accelerated solvent extraction/gel permeation chromatography and solid phase extraction/gas chromatography-mass spectrometry [J]. Chinese Journal of Chromatography, 2008, 26(1): 22-28
[8] LIU Hong-mei, KONG Wei-jun, Gong Bao, et al. Rapid analysis of multi-pesticides in morinda officinalis by gc-ecd with accelerated solvent extraction assisted matrix solid phase dispersion and positive confirmation by GC-MS [J]. Journal of Chromatography B., 2015, 974(1): 65-74
[9] CAI Ling-shuang, XING Jun, DONG Li, et al. Application of polyphenyl-methylsiloxane coated fiber for solid-phase microextraction combined with microwave-assisted extraction for the determination of organochlorine pesticides in chinese teas [J]. J. Chromatogr. A, 2003, 1015(1-2, 10): 11-21
[10] LI Bin, ZENG Fan-gang, DONG Qi-chun, et al. Rapid determination method for 12 pyrethroid pesticide residues in tea by stir bar sorptive extraction-thermal desorption-gas chromatography [J]. Physics Procedia, 2012, 25: 1776-1780
[11] WANG Xiu-hong, CHENG Jing, ZHOU Hong-bin, et al. Development of a simple combining apparatus to perform a magneticstirring-assisted dispersive liquid-liquid microextraction and its application for the analysis of carbamate and organophosphorus pesticides in tea drinks [J]. Analytica Chimica Acta, 2013, 787(17): 71-77[12] LIU Dan, MIN Shun-geng. Rapid analysis of organochlorine and pyrethroid pesticides in tea samples by directly suspended droplet microextraction using a gas chromatography-electron capture detector [J]. J. Chromatogr. A, 2012, 1235(27): 166-173
[13] Ana Lozano, Łukasz Rajski, Noelia Belmonte-Valles, et al. pesticide analysis in teas and chamomile by liquid chromatography and gas chromatography tandem mass spectrometry using a modified QuEChERS method: validation and pilot survey in real samples [J]. J. Chromatogr. A, 2012, 1268(14): 109-122
[14] Silvia de Sousa Freitas, Fernando Mauro Lanças. Matrix effects observed during pesticides residue analysis in fruits by GC [J]. J. Sep. Sci., 2009, 32(21): 3698-705
[15] ZHAO Dong-dong, LIU Xiao-yu, SHI Wang-rong, et al. Determination of cypermethr in residuesin crucian carp tissues by MSPD/GC-ECD [J]. Chromatographia, 2011, 73: 1021-1025
[16] 林竹光,金珍,刘勇,等.GC-NCI-MS分析茶叶中17种有机氯和拟除虫菊酯农药残留[J].高等学校化学学报,2005,26 (12):2218-2222 LIN Zhu-guang, JIN Zhen, LIU Yong, et al. Multiresidue determination of 17 organochlorine and pyrethroid pesticides in tea by gas chromatography-negative chemical ionizationmass spectrometry [J]. Chem. J. Chinese Universities, 2005, 26(12): 2218-2222
[17] HU Yan-yun, ZHENG Ping, HE You-zhao, et al. Response surface optimization for determination of pesticide multiresidues by matrix solid-phase dispersion and gas chromatography [J]. J. Chromatogr. A, 2005, 1098(1-2): 188-193
Development of Simple Sample Pretreatment Method for the Simultaneous Analysis of Organochlorine and Pyrethroid Pesticide Residues in Tea
Abstract:Eleven types of organochlorine and pyrethroid pesticides in tea samples were analyzed simultaneously and rapidly in this study. Using a simple pretreatment method, the tea samples were fully ground with the adsorbent, and then the matrix was purified by column chromatography followed by detection using gas chromatography (GC). Adsorbent, column, and eluent conditions were investigated, and the optimized conditions were as follows: 1.0 g of sample immersed in water was combined completely with 4.0 g of neutral aluminum oxide, and the mixture was transferred into a self-made glass column containing 2.0 g of anhydrous sodium sulfate, 2.0 g of Florisil, and 0.1 g of activated carbon for purification and pre-separation. The combined sample was then eluted with 20 mL of ethyl acetate and n-hexane (2:3, V:V). For this method, the linear range of the calibration curves was between 0.1 mg/L and 2.0 mg/L, the coefficients of determination were ≥0.9962 for all target analytes except cyfluthrin and cypermethrin, and the limit of detection ranged from 0.0000321 ng to 0.1451379 ng. The recoveries ranged from 68.44% to 119.20%, and the relative standard deviations of the recovery results at different spiking levels were lower than 14.73%. In addition, the matrix effect (ME) issue in the tea sample was investigated in detail, and the method of matrix-matched calibration was applied to solve the ME issue effectively. This developed method is fast, accurate, simple, and environmentally friendly, and is suitable for the simultaneous and rapid separation and analysis of various pesticide residues in tea.
Key words:analysis of pesticide residues; tea; sample pretreatment method, matrix effects
文章篇号:1673-9078(2017)2-223-229
DOI:10.13982/j.mfst.1673-9078.2017.2.034
收稿日期:2016-02-15
基金项目:国家自然科学基金资助项目(31471706);园艺学优势特色重点学科开放基金重点项目(2014YYX006)